
Idiopathic Pulmonary Fibrosis

Newly diagnosed with IPF? Start here.
If you're newly diagnosed with idiopathic pulmonary fibrosis, we recommend you begin your learning journey with our in-depth information series, PF Basics: Info for newly diagnosed patients.
Right now, the most important thing we want you to remember is that you’re not alone. The PFF is here for you at every stage of your journey. We can also connect you with other people who have this disease so that you can feel supported and heard.
If you, your family members, or your friends have questions, please call the PFF Help Center at 1.844.825.5733 or email us at help@pulmonaryfibrosis.org.
Continue scrolling and reading through this page for a brief overview of IPF. For a more in-depth overview, refer to PF Basics: Info for newly diagnosed patients.
What is Idiopathic Pulmonary Fibrosis?
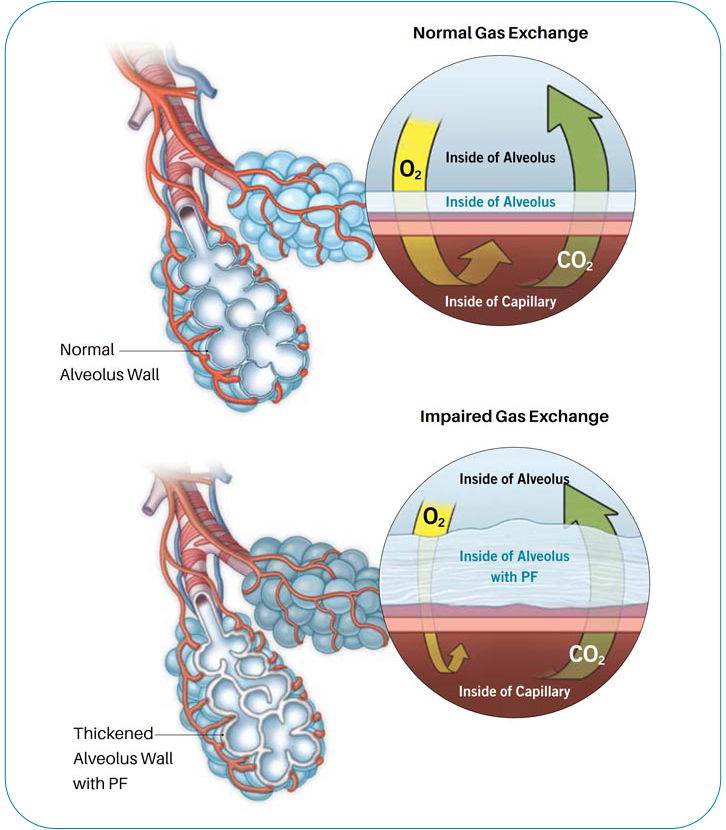
There are more than 200 types of interstitial lung diseases (ILD), which are characterized by varied amounts of inflammation, scarring, or both, that damage the ability of the lung to absorb oxygen from the air. Pulmonary fibrosis (PF) means scarring of the lung and can be seen in many types of ILD.
When a person is diagnosed with PF, sometimes a doctor is able to find the cause of the disease. In many cases a doctor can’t find a reason why a person has developed pulmonary fibrosis. When the cause of the disease is not known, the fibrosis may be termed “idiopathic."
Idiopathic pulmonary fibrosis (IPF) is the most common type of idiopathic interstitial lung disease. But there are multiple forms of ILD that are idiopathic. Your doctor will use detailed X-rays of your lungs called high-resolution computed tomography (HRCT) and sometimes a lung biopsy to look for a specific pattern of scarring on your lungs, called usual interstitial pneumonia (UIP). A diagnosis of IPF requires that your doctor cannot find a cause and the presence of a pattern of UIP on either HRCT or a surgical lung biopsy sample.
More than 250,000 Americans are living with PF and ILD. Idiopathic pulmonary fibrosis is one of the most common forms of PF. The prevalence of PF and ILD is on the rise with
more than 50,000 new cases diagnosed annually.

What Are the Risk Factors for IPF?
Although the word “idiopathic” means “of unknown cause,” we know that there are certain factors that increase the risk of receiving an IPF diagnosis. Current and former smokers are more likely to develop IPF than those who have never smoked. Age may play a role because IPF is rare before the age of 50.
A family history of pulmonary fibrosis is also a risk factor, as are certain genes, such as MUC5B, TERT, TERC, DKC1, RTEL1, AKAP13, DSP, FAM13A, DPP9, and TOLLIP. Some evidence suggests that certain viral infections, air pollution, and some exposures in the workplace may also be risk factors for IPF.
There are also conditions, such as gastroesophageal reflux disease (acid reflux, heartburn, or GERD), sleep apnea, or pulmonary hypertension (PH) that are often present in people who are diagnosed with IPF. These conditions are called “comorbid conditions” or “comorbidities.” The presence of risk factors and comorbidities provide clues that may be helpful to a physician who suspects that an ILD is present in a patient.
What Are the Symptoms of IPF?
IPF shares the same general symptoms as other forms of pulmonary fibrosis. Early in the course of the disease, many people with IPF have no symptoms. Some might have a bothersome cough, or notice that they tire more easily. As the disease progresses and scarring builds up in the air sacs of the lungs, breathlessness during exercise and daily activities becomes common. People living with PF may also feel fatigued, anxious, or depressed.

How is IPF Diagnosed?
When a doctor or other healthcare provider suspects that a patient has ILD, they will collect information about the patient’s medical and personal history, work and home environment, hobbies, and illness that may be present in the family. This can help a doctor identify exposures or other diseases that might have caused lung injury and scarring. The doctor will also often order pulmonary function tests, a chest x-ray, blood work, and a high-resolution CT scan.
Pulmonary function tests measure how much air the lungs can hold, and how the lungs are working overall. Scarring can cause the lungs to shrink, and it can also make them stiff and unable to fully expand. This means the lungs are able to hold less air. Scar tissue may also affect the ability of the lungs to transfer oxygen to the bloodstream.
Blood work (serology) can provide information about exposures that may have caused a person’s lung injury, or show that other diseases are present which may be associated with the development of PF. If a cause for the PF, such as an autoimmune disease or environmental exposure, is identified, then the diagnosis is not IPF.
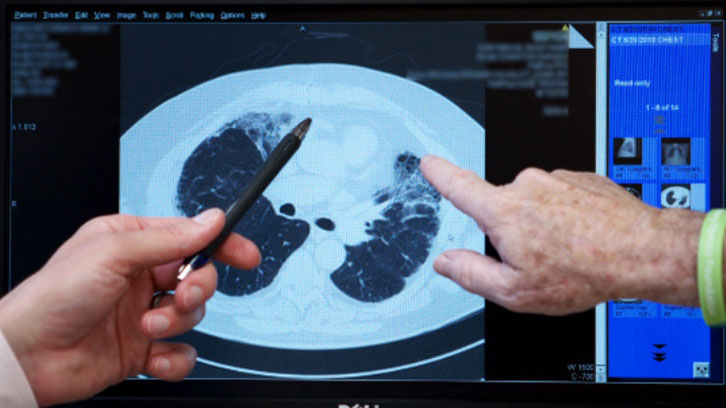
A high-resolution computed tomography (HRCT) scan is a special type of x-ray that shows fine detail of the lung tissue. On a CT scan (also known as CAT scan) healthy lung tissue looks nearly black, and scar tissue and inflammation appear grey or white. A specific scarring pattern called usual interstitial pneumonia (UIP), can help to diagnose IPF when it is seen on an HRCT scan.
After reviewing the results of all of these tests, a physician can often confirm a diagnosis of IPF. Sometimes more information is needed and a lung biopsy may be performed.
If a Cause Can’t be Found for My Pulmonary Fibrosis, Then is My Diagnosis IPF?
Diagnosing an ILD involves many different types of information, and making an accurate diagnosis can be complicated. Some patients’ disease is easier to diagnose because the cause and type of disease are clear. For other patients a cause cannot be found, and they may not fit easily into any particular category of the disease. In addition to IPF, there are many ILDs of unknown cause – meaning that there are many “idiopathic” ILDs other than IPF. These idiopathic ILDs and their abbreviations can be confusing. This chart shows these different types of idiopathic ILDs.


How is IPF Treated?
Supplemental oxygen, pulmonary rehabilitation, and management of symptoms are important treatment options for many types of pulmonary fibrosis, depending on how severe the disease is. Smoking cessation and routine vaccinations are important parts of living with pulmonary fibrosis. Lung transplantation may be an appropriate treatment for some people living with IPF.
What is My Prognosis?
Idiopathic pulmonary fibrosis is a progressive disease, which means that fibrosis builds up over time, gradually causing an increase in breathlessness and the need for increasing amounts of oxygen. The progression of IPF is variable, with some people experiencing slower but steady decline in lung function and others developing more rapidly worsening disease. Patients with IPF may have sudden declines in functioning, known as acute exacerbations. Eventually, lung failure (medically called “respiratory failure”) can develop, which is a life-threatening condition.
It is important for you to know that there is no way to predict how long someone with IPF will live. You may have heard that the average survival of people living with IPF is only “three to five years.” This is an outdated statistic. With earlier diagnosis and better treatments available, many people live much longer than three to five years. Others will develop respiratory failure sooner than three years, with some becoming very ill within months or just a few years after their diagnosis. No one can predict exactly how long you will live with IPF. Everyone is different. Your doctor can give you more detailed information about your prognosis.


Are There Experimental Therapies Available?
Some patients choose to participate in clinical trials, which are research studies that explore whether a medical strategy, treatment, or device is safe and effective for use in humans. These studies may also show which medical approaches work best for
certain illnesses or groups of people. The purpose of clinical trials is research, so the studies follow strict scientific standards that protect patients and help produce reliable study results. Participation in clinical studies by people living
with PF is critical so that we can learn more about the causes of pulmonary fibrosis and find new treatments for PF.
You can learn more about clinical trials in the PFF Clinical Trials Education Center.
What Should I Do Next?
If you have been recently diagnosed with pulmonary fibrosis, consider making an appointment with a pulmonologist who has experience caring for patients with PF. A knowledgeable team of experts will help make sure you receive an accurate diagnosis and
the most up-to-date treatments and management recommendations. To assist you in finding pulmonologists nearest to your home who provide expert care to patients with PF, the Pulmonary Fibrosis Foundation established the PFF Care Center Network, which includes 86 medical centers throughout the United States. You can search for the PFF Care Center Network medical center closest to
you here.
We also recommend that you join a pulmonary fibrosis support group. Connecting with other individuals facing the same illness can help you and your family feel less alone in your journey with pulmonary fibrosis. Support groups can supplement the care
you receive from your healthcare team by providing emotional support and education. You can find a list of support groups here.
Support groups can help those living with pulmonary fibrosis to:
- Learn about their disease and available treatments
- Feel supported by others who share similar experiences living with PF
- Learn to navigate the healthcare system more effectively
- Improve coping skills, connect with helpful resources, and more
Contact the PFF Help Center by calling 844.TalkPFF (844.825.5733) or email help@pulmonaryfibrosis.org. We can help answer your questions, discuss your concerns, and connect you with resources.


What researchers are doing to help
The Pulmonary Fibrosis Foundation is leading the fight by funding promising research and we need your help.
One of our key research programs is the PFF Community Registry. We invite eligible participants to join by completing a series of simple surveys. Your survey responses will be used by researchers to better understand how PF and ILD progress over time, respond to treatments, and how the diseases affect individuals. The more individuals who join and provide responses, the closer we come to a cure.
Eligible participants include:
- Patients living with PF and ILD
- Lung transplant recipients who have had PF or ILD
- Caregivers and biological family members of patients with PF or ILD, including those who have passed away
Enrolling in the PFF Community Registry is entirely online. All you need is internet access. Learn more about this groundbreaking program by visiting the PFF Community Registry homepage.
Educational Materials
Find reliable information and trusted resources that can help you learn about pulmonary fibrosis and live better with PF.
-
View Full Details
Pulmonary Fibrosis Information Guide
Our comprehensive guide provides reliable information about pulmonary fibrosis, the diagnostic process, treatment options, and more. -
View Full Details
Idiopathic Pulmonary Fibrosis Fact Sheet
This fact sheet provides an overview of idiopathic pulmonary fibrosis, symptoms, causes, diagnosis, and much more.

PFF Help Center
For those living with pulmonary fibrosis, obtaining the most accurate and current information can be a frustrating and challenging task. Let us help you find your answers.